What we do
We use systems biology approaches to uncover the underlying principles governing the operation of genetics networks. Specifically, we integrate computational modeling and data analysis to elucidate the relationship among robustness of network dynamics, stochasticity in gene expression and heterogeneity in a cell population. Our studies will contribute to a systems-level understanding of biological processes determining cellular state transitions.
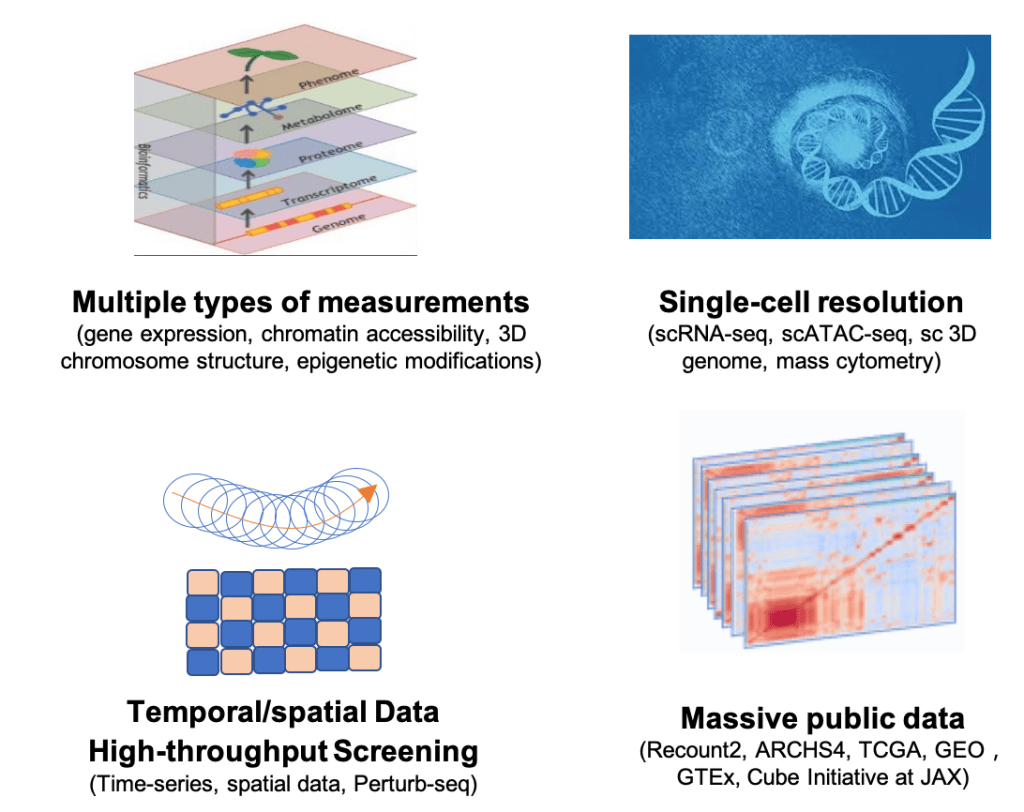
New advances in genomics provide new opportunities for Systems Biology modeling:
With the new advances in technologies, researchers can not only measure multiple types of genomics data at the same time, but also at single-cell resolution, with temporal and spatial information. Most importantly, these data are made publicly available almost immediately.
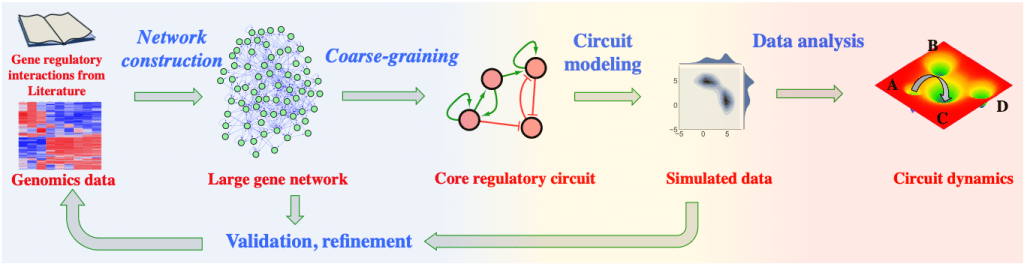
Overall Strategy
We first integrate both literature and genomics data to construct a large gene network with bioinformatics. Then using mathematical modeling, we aim to identify a core gene regulatory circuit. Mathematical modeling can also simulate circuit dynamics, from which we propose new predictions that can be tested experimentally. This is an iterative process to improve circuit models.
Systems biology modeling
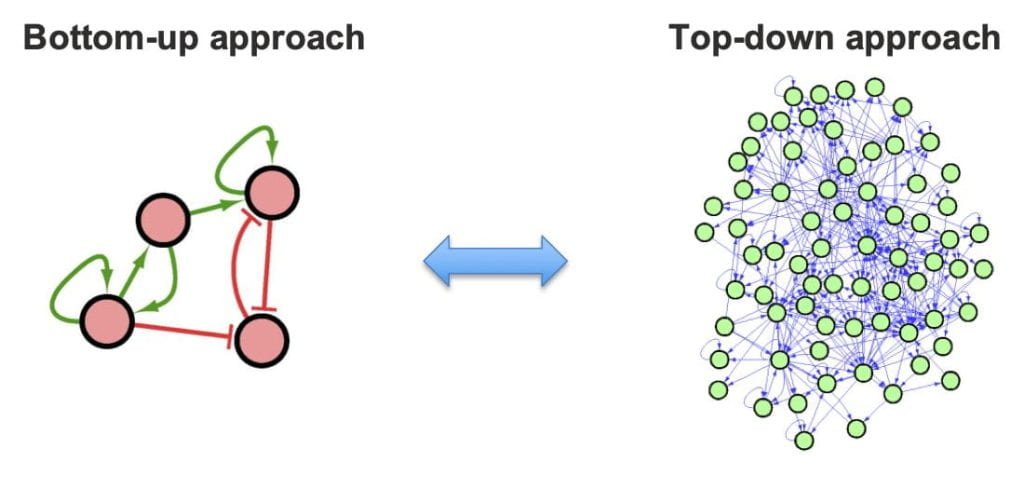
Integrating bottom-up and top-down Systems Biology The key is a robust and powerful mathematical modeling method. There are many challenges, including the following issues. (1) Unlike synthetic circuits, which can be designed as isolated as possible to the host, biological circuits can be highly coupled, making it hard to find circuits of the right size to model. (2) A popular way to model a circuit is to use rate equations. However, most of the required kinetic parameters are not directly measurable, especially in vivo. Thus, one has to make either educated guess or fit the parameters against data such as gene expression. This becomes an issue for large circuits with large number of parameters, making this approach prone to overfitting.
This parameter problem is a long-standing issue in almost every systems biology studies. Like this famous saying, “with four parameters I can fit an elephant, and with five I can make him wiggle his trunk.”
Reference:
A. Katebi, D. Ramirez, M. Lu. (2021) Computational systems-biology approaches for modeling gene networks driving epithelial–mesenchymal transitions. Comput. Syst. Oncol. 1(2):e1021
RACIPE
To address these questions, we developed a new method, named random circuit perturbation (RACIPE), to address the parameter issue. Unlike the traditional approach, where we focus on a fixed parameter set. Here in RACIPE, we generate an ensemble of models with random kinetic parameters. We then use numerical methods to analyze the dynamics of each model in a high throughput way, so that we can perform statistical analysis on these models to find the most robust behaviors. RACIPE is different from some studies where the parameter space is explored to find best fitting. Instead, we consider each model is valid and corresponds to the same circuit in different situations, such as signaling states and epigenetic states We designed a sampling scheme to cover the parameter space.
We first applied RACIPE to simple toggle-switch-like circuit motifs. In each case, we found that the stable steady states from a large ensemble of random models form robust clusters, corresponding to the states in which the circuit operates. For example, a toggle switch (TS) allows two robust states; a TS with two-sided self-activations allows additional state with hybrid expression. For circuits with coupled TSs, each additional TS opens a new state — a feature that can also been observed from Principle Component Analysis of the RACIPE-generated gene expression. We also applied RACIPE to a large EMT gene regulatory circuit, from which we predicted two hybrid EMT states (EM1 and EM2) in addition to epithelial and mesenchymal states.
Reference:
B. Huang*, M. Lu*, D. Jia, E. Ben-Jacob, H. Levine, J. Onuchic. (2017) Interrogating the topological robustness of gene regulatory circuits by randomization. PLoS Comput Biol.13(3):e1005456 (*equal contribution)
sRACIPE
Stochasticity in gene expression impacts the dynamics and functions of gene regulatory circuits. Intrinsic noises, including those that are caused by low copy number of molecules and transcriptional bursting, are usually studied by stochastic simulations. However, the role of extrinsic factors, such as cell-to-cell variability and heterogeneity in the microenvironment, is still elusive. To evaluate the effects of both the intrinsic and extrinsic noises, we develop a method, named sRACIPE, by integrating stochastic analysis with random circuit perturbation (RACIPE) method. In sRACIPE we develop two stochastic simulation schemes, aiming to reduce the computational cost without sacrificing the convergence of statistics. One scheme uses constant noise to capture the basins of attraction, and the other one uses simulated annealing to detect the stability of states. By testing the methods on several synthetic gene regulatory circuits and an epithelial–mesenchymal transition network in squamous cell carcinoma, we demonstrate that sRACIPE can interpret the experimental observations from single-cell gene expression data. We observe that parametric variation (the spread of parameters around a median value) increases the spread of the gene expression clusters, whereas high noise merges the states. Our approach quantifies the robustness of a gene circuit in the presence of noise and sheds light on a new mechanism of noise-induced hybrid states.
Reference:
V. Kohar and M. Lu. (2018) Role of noise and parametric variation in the dynamics of gene regulatory circuits. NPJ Syst. boil. Appl. 4: 40
GeneEx
GeneEx is an interactive web-app that employs an ODE-based mathematical modeling approach to simulate, visualize, and analyze gene regulatory circuits (GRCs) for an explicit kinetic parameter set or for a large ensemble ofrandom parameter sets. GeneEx offers users the freedom to modify many aspects of the simulation such as the parameter ranges, the levels of gene expression noise, and the GRC network topology itself. This degree of flexibility allows users to explore a variety of hypotheses by providing insight into the number and stability of attractors for a given GRC. Moreover, users have the option to upload, and subsequently compare, experimental gene expression data to simulated data generated from the analysis of a built or uploaded custom circuit. Finally, GeneEx offers a curated database that contains circuit motifs and known biological GRCs to facilitate further inquiry into these. Overall, GeneEx enables users to investigate the effects of parameter variation, stochasticity, and/or topological changes on gene expression for GRCs using a systems-biology approach.
Reference:
V. Kohar, D. Gordin, A. Ketabi, M. Lu. (2021) Gene Circuit Explorer (GeneEx): an interactive web-app and database for visualizing, simulating and analyzing gene regulatory circuits. Bioinformatics. 37(9):1327-29
SacroGraci – Network coarse-graining
One major challenge in systems biology is to understand how various genes in a gene regulatory network (GRN) collectively perform their functions and control network dynamics. This task becomes extremely hard to tackle in the case of large networks with hundreds of genes and edges, many of which have redundant regulatory roles and functions. The existing methods for model reduction usually require the detailed mathematical description of dynamical systems and their cor- responding kinetic parameters, which are often not available. Here, we present a data-driven method for coarse-graining large GRNs, named SacoGraci, using ensemble-based mathematical modeling, dimensionality reduction, and gene cir- cuit optimization by Markov Chain Monte Carlo methods. SacoGraci requires network topology as the only input and is robust against errors in GRNs. We benchmark and demonstrate its usage with synthetic, literature-based, and bioin- formatics-derived GRNs. We hope SacoGraci will enhance our ability to model the gene regulation of complex biological systems.
Reference:
C. Caranica, and M. Lu. (2023) A Data-Driven Optimization Method for Coarse-Graining Gene Regulatory Networks. iScience. 26(2): 105927
Topological motifs and their coupling in gene circuits
One of the major challenges in biology is to understand how gene interactions collaborate to determine overall functions of biological systems. Here, we present a new computational framework that enables systematic, high- throughput, and quantitative evaluation of how small transcriptional regulatory circuit motifs, and their coupling, contribute to functions of a dynamical biological system. We illustrate how this approach can be applied to identify four-node gene circuits, circuit motifs, and motif coupling responsible for various gene expression state distributions, including those derived from single-cell RNA sequencing data. We also identify seven major classes of four-node circuits from clustering analysis of state distributions. The method is applied to establish phenomenological models of gene circuits driving human neuron differentiation, revealing important biologically relevant regulatory interactions. Our study will shed light on a better understanding of gene regulatory mechanisms in creating and maintaining cellular states.
Reference:
B. Clauss, and M. Lu. (2023) A Quantitative Evaluation of Topological Motifs and Their Coupling in Gene Circuit State Distributions. iScience. 26(2): 106029L. Huang, B. Clauss, M. Lu. (2022) What Makes a Functional Gene Regulatory Network? A Circuit Motif Analysis. J. Phys. Chem. B. 126, 10374–10383

Research Paper/Protocol Example
Highlight one of your protocols or a relevant publication. Add an image or figure to the left and a link below.
Cell state Transitions
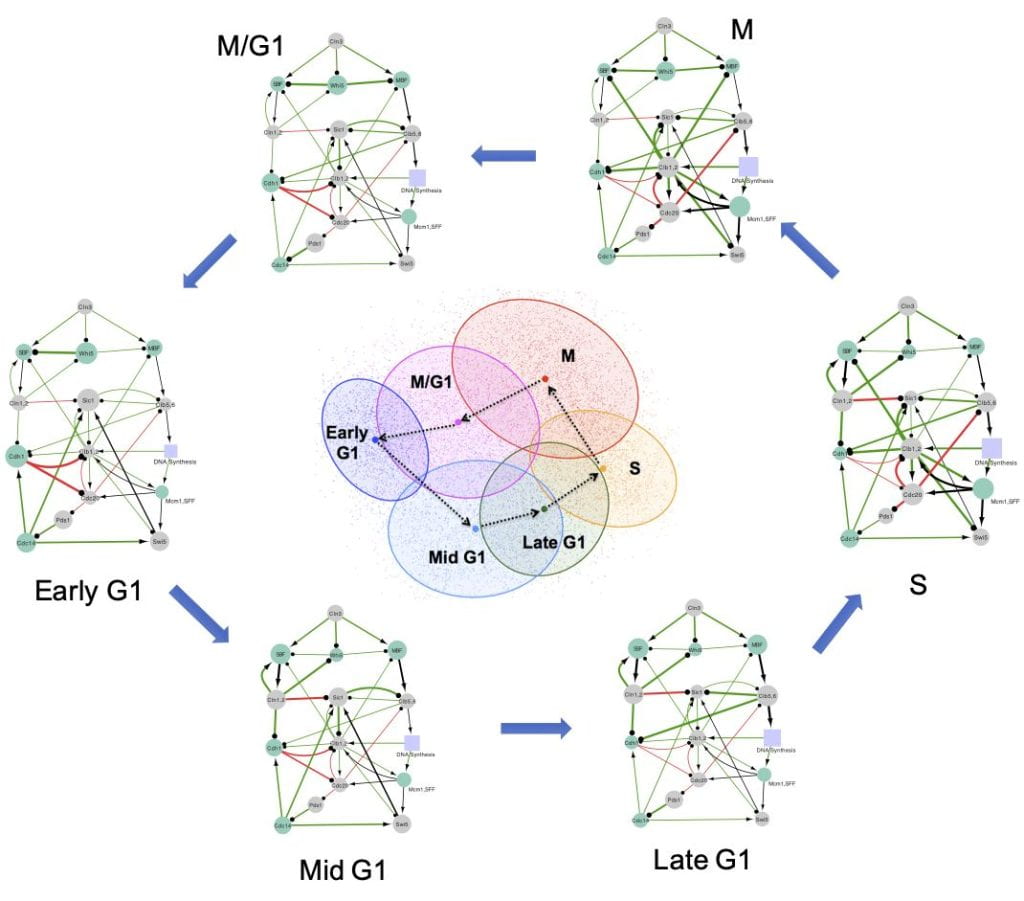
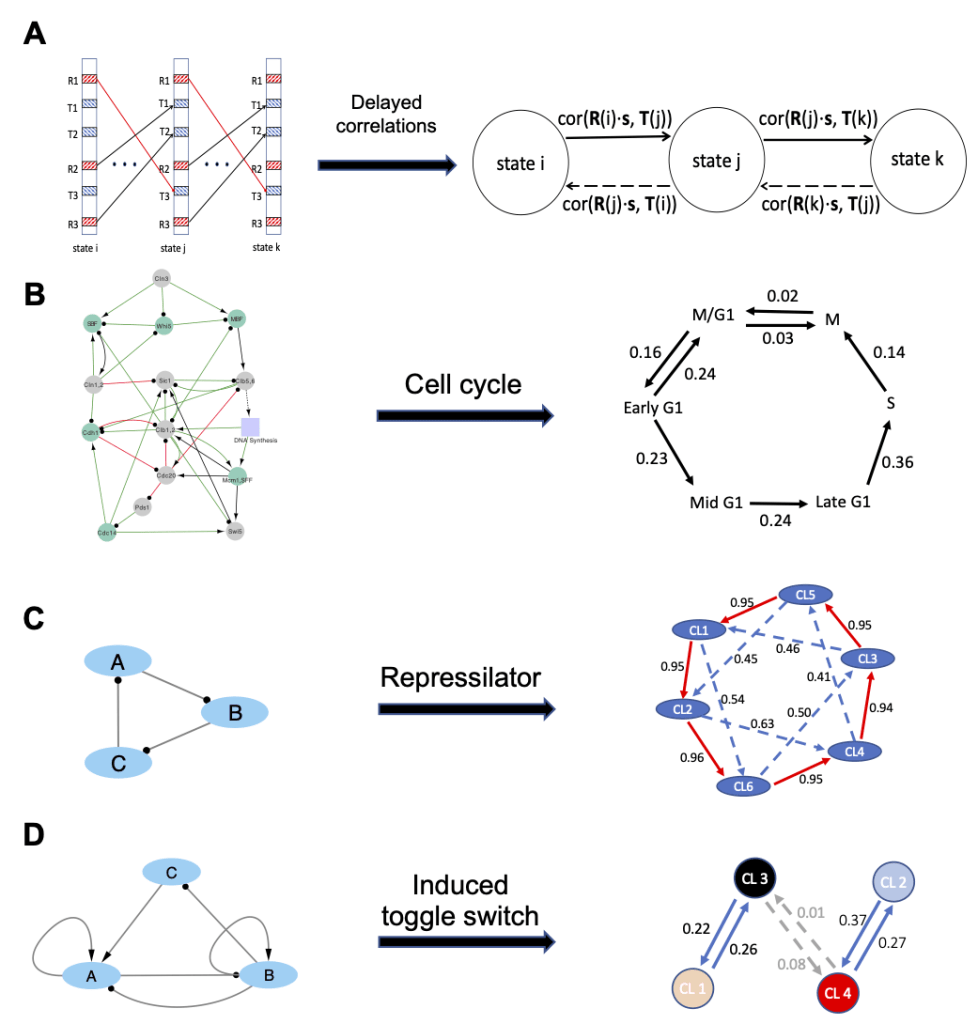
Many biological processes involve precise cellular state transitions controlled by complex gene regulation. Here, we use budding yeast cell cycle as a model system and explore how a gene regulatory circuit encodes essential information of state transitions. We present a generalized random circuit perturbation method for circuits containing heterogeneous regulation types and its usage to analyze both steady and oscillatory states from an ensemble of circuit models with random kinetic parameters. The stable steady states form robust clusters with a circular structure that are associated with cell cycle phases. This circular structure in the clusters is consistent with single-cell RNA sequencing data. The oscillatory states specify the irreversible state transitions along cell cycle progression.
Furthermore, we define a metric based on delayed correlation to infer state transition patterns between states directly from stable steady states. The method has been applied to the cell cycle circuit, the repressilator circuit, and an induced toggle switch circuit.
Reference:
A. Katebi, V. Kohar, M. Lu. (2020) Random Parametric Perturbations of Gene Regulatory Circuit Uncover State Transitions in Cell Cycle. iScience. 23(6): 101150
Random Parametric Perturbations of Gene Regulatory Circuit Uncover State Transitions in Cell Cycle.
Highlight one of your protocols or a relevant publication. Add an image or figure to the right and a link below.
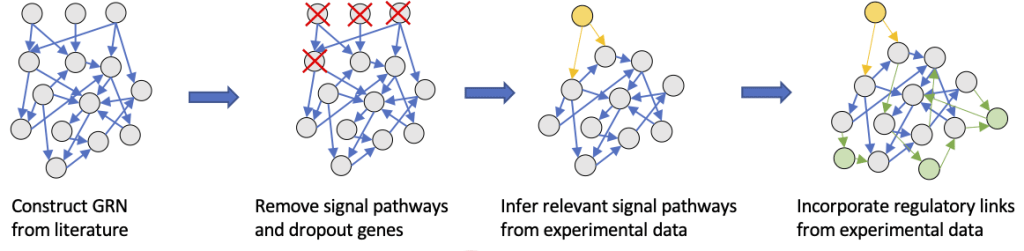
Network Construction
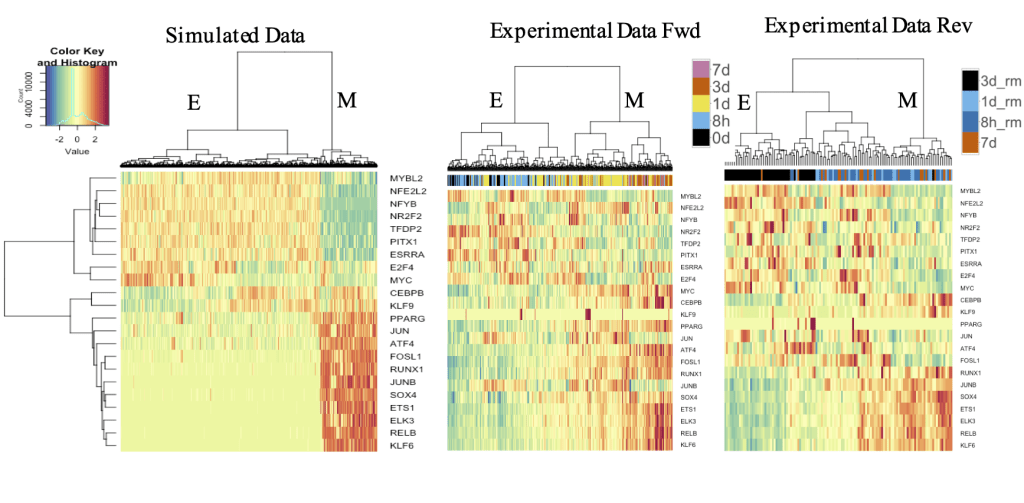
Toward Modeling Context-Specific EMT Regulatory Networks Using Temporal Single Cell RNA-Seq Data
Many previous studies have been conducted to model gene regulatory circuits (GRCs) using interactions from the literature. While this approach can depict the generic regulatory interactions, it falls short of capturing context-specific features. Here, we explore the effectiveness of a combined bioinformatics and mathematical modeling approach to construct context-specific GRCs directly from transcriptomics data. Using time-series single cell RNA-sequencing data from four different cancer cell lines treated with three EMT-inducing signals, we identify context-specific activity dynamics of common EMT transcription factors. In particular, we observe distinct paths during the forward and backward transitions, as is evident from the dynamics of major regulators such as NF-KB (e.g., NFKB2 and RELB) and AP-1 (e.g., FOSL1 and JUNB). For each experimental condition, we systematically sample a large set of network models and identify the optimal GRC capturing context-specific EMT states using a mathematical modeling method named Random Circuit Perturbation (RACIPE). The results demonstrate that the approach can build high quality GRCs in certain cases, but not others and, meanwhile, elucidate the role of common bioinformatics parameters and properties of network structures in determining the quality of GRCs. We expect the integration of top-down bioinformatics and bottom-up systems biology modeling to be a powerful and generally applicable approach to elucidate gene regulatory mechanisms of cellular state transitions.
Reference:
D. Ramirez, V. Kohar, M. Lu. (2020) Toward Modeling Context-Specific EMT Regulatory Networks Using Temporal Single Cell RNA-Seq Data. Front Mol Biosci. 7:54
NetAct
A major question in systems biology is how to identify the core gene regulatory circuit that governs the decision-making of a biological process. Here, we develop a computational platform, named NetAct, for constructing core transcription factor regulatory networks using both transcriptomics data and literature-based transcription factor-target databases. NetAct robustly infers regulators’ activity using target expression, constructs networks based on transcriptional activity, and integrates mathematical modeling for validation. Our in-silico benchmark test shows that NetAct outperforms existing algorithms in inferring transcriptional activity and gene networks. We illustrate the application of NetAct to model networks driving TGF-β-induced epithelial-mesenchymal transition and macrophage polarization.
Reference:
K. Su, A. Katebi, V. Kohar, B. Clauss, D. Gordin, Z.S. Qin, R.K.M. Karuturi, S. Li, M. Lu. (2022) NetAct: a computational platform to construct core transcription factor regulatory networks using gene activity. Genome Biology. 23, 270

Research Paper/Protocol Example
Highlight one of your protocols or a relevant publication. Add an image or figure to the left and a link below.
Cancer Networks
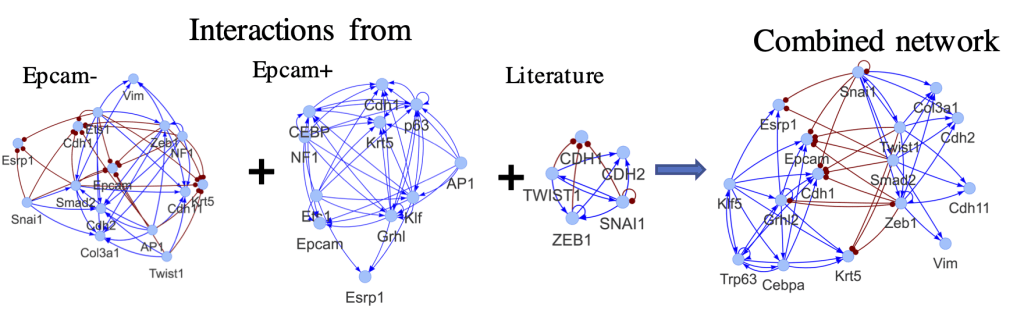
We construct an EMT transcriptional regulatory circuit by combining existing Epcam− and Epcam+ networks in squamous cell carcinoma (SCC) and the other literature data. The activating links are shown as blue lines and arrows, and the inhibitory links are shown as red lines and dots.
Reference:
V. Kohar and M. Lu. (2018) Role of noise and parametric variation in the dynamics of gene regulatory circuits. NPJ Syst. boil. Appl. 4: 40
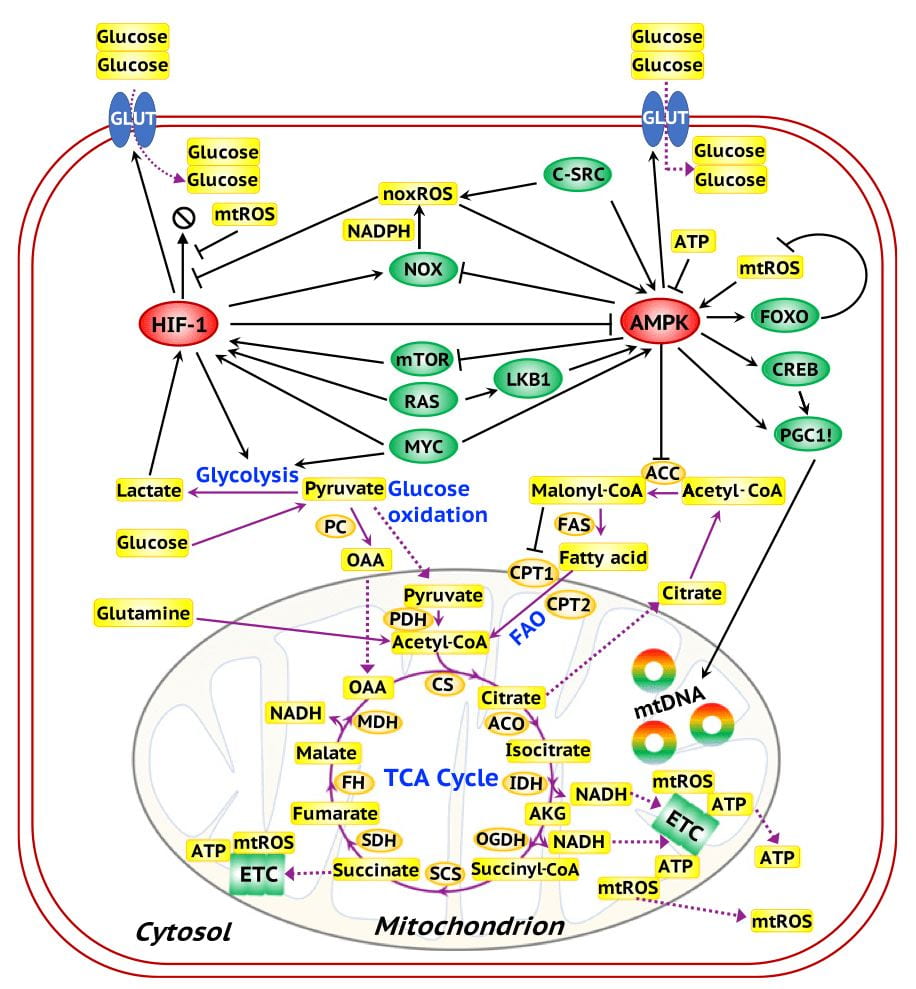
A comprehensive regulatory network of glycolysis and OXPHOS. The network is composed of the master regulators AMPK and HIF-1 (red ovals), downstream target genes of the master regulators and oncogenes (green ovals), the enzyme genes (orange ovals), and metabolites (yellow rectangles). Black arrows represent excitatory regulation and black bar-headed arrows represent inhibitory regulation. Purple solid lines represent the chemical reactions in metabolic pathways and purple dotted lines represent the transportation of metabolites.
Reference:
D. Jia, M. Lu, K.H. Jung, J.H. Park, L. Yu, J. Onuchic, B.A. Kaipparettu, H. Levine. (2019) Elucidating Cancer Metabolic Plasticity by Coupling Gene Regulation with Metabolic Pathways, Proc. Natl. Acad. Sci. U.S.A.116(9): 3909-18
Research Paper/Protocol Example
Highlight one of your protocols or a relevant publication. Add an image or figure to the right and a link below.